
Il dlgs 20 febbraio 2026, numero 30, che recepisce la Direttiva (UE) 2024/825, stabilisce che le asserzioni ambientali ingannevoli sono pratiche commerciali scorrette: il decreto si applica a tutti i prodotti destinati ai consumatori, compresi i dispositivi medici, settore nel quale i green claim stanno emergendo con forza crescente. Ricapitoliamo allora brevemente le regole sulla pubblicità dei dispositivi medici, per poi analizzare come il nuovo decreto può impattare in questo settore.
La pubblicità dei dispositivi medici al consumatore: il quadro normativo vigente
La pubblicità dei dispositivi medici rivolta al pubblico è oggi disciplinata da un sistema normativo stratificato, che opera su tre livelli distinti ma tra loro interconnessi.
- Il livello europeo. L’articolo 7 del Regolamento (UE) 2017/745 (MDR) ha introdotto una norma ad hoc per le “dichiarazioni” (o claims) relative ai dispositivi medici. La disposizione vieta, in qualsiasi forma di comunicazione – etichettatura, istruzioni per l’uso, messa a disposizione, messa in servizio e pubblicità – il ricorso a testi, denominazioni, marchi, immagini e segni figurativi o di altro tipo che possano indurre l’utilizzatore o il paziente in errore per quanto riguarda la destinazione d’uso, la sicurezza e le prestazioni del dispositivo. L’ambito di applicazione dell’articolo 7 MDR è particolarmente ampio. Il divieto copre qualsiasi mezzo di comunicazione – diretta (testi) o indiretta (denominazioni, marchi, immagini, segni figurativi) – idoneo a indurre in errore il soggetto a cui ci si rivolge. Si noti che il MDR utilizza la nozione di “induzione in errore”, un istituto giuridico tipico della disciplina della pubblicità ingannevole (articolo 2, comma 1, lett. b), D.Lgs. 145/2007) e delle pratiche commerciali sleali (articolo 21, comma 1, del Codice del Consumo).
- Il livello nazionale. L’articolo 26 del dlgs 5 agosto 2022, numero 137, di adeguamento dell’ordinamento italiano al MDR, ha ripreso e aggiornato la disciplina della pubblicità dei dispositivi medici. Al primo comma la norma prevede il divieto di pubblicità verso il pubblico per alcune categorie di dispositivi: quelli su misura, quelli che richiedono obbligatoriamente l’assistenza di un medico o di un professionista sanitario e quelli la cui vendita al pubblico è subordinata a prescrizione medica. Per tutti i dispositivi medici diversi da quelli appena elencati, la pubblicità verso il pubblico è consentita, ma – ferme restando le prescrizioni dell’articolo 7 MDR – è soggetta ad autorizzazione del Ministero della Salute (articolo 26, comma 3, dlgs 137/2022). Le modalità operative sono disciplinate dal decreto del Ministero della Salute del 26 gennaio 2023, che individua anche le fattispecie per le quali non è necessaria l’autorizzazione ministeriale. Preme precisare che l’articolo 26 del dlgs 137/2022 si applica esclusivamente ai messaggi aventi natura pubblicitaria, non a tutte le fattispecie di comunicazione regolate dall’articolo 7 MDR (quali etichettatura, istruzioni per l’uso, messa a disposizione, messa in servizio).
- Il Codice del Consumo e la competenza dell’AGCM. A questo doppio livello di regolazione – europeo e settoriale nazionale – si affianca la disciplina generale sulle pratiche commerciali scorrette contenuta nel Codice del Consumo (articoli 18-27-quater, dlgs 206/2005). Il punto è cruciale: la pubblicità dei dispositivi medici al consumatore non è soggetta soltanto al regime autorizzatorio del Ministero della Salute. Essa rientra a pieno titolo anche nell’ambito di applicazione del Codice del Consumo e, di conseguenza, nella competenza sanzionatoria dell’AGCM. Questo principio è stato affermato dall’articolo 27, comma 1-bis, del Codice del Consumo (introdotto dal D.Lgs. 21/2014), secondo cui “anche nei settori regolati […] la competenza ad intervenire nei confronti delle condotte dei professionisti che integrano una pratica commerciale scorretta […] spetta, in via esclusiva, all’Autorità garante della concorrenza e del mercato” e successivamente chiarito dal Consiglio di Stato 29.11.2018 numero 6795 con la quale si dichiara che le discipline speciali dei farmaci e dispositivi medici con complementare con quelle del Codice del Consumo, confermando quindi la competenza dell’AGCM a valutare la scorrettezza di una pratica commerciale purché la valutazione sia condotta alla luce dei criteri generali degli articoli 20‑27 Codice del consumo.
Ne deriva un quadro a doppio binario: il fabbricante di dispositivi medici che viola le regole sulla pubblicità può essere sanzionato sia dal Ministero della Salute (ai sensi dell’articolo 27, comma 5, dlgs 137/2022, con sanzione da 20 mila a 120 mila euro per pubblicità non autorizzata) sia dall’AGCM (ai sensi dell’articolo 27, comma 9, del Codice del Consumo, con sanzione fino a 10.000.000 di euro, o fino al 4% del fatturato annuo nei casi più gravi).
È in questo contesto che si inserisce il dlgs 30/2026. Le nuove fattispecie di greenwashing introdotte dal decreto diventano automaticamente parte del Codice del Consumo e, dunque, si applicano anche alla pubblicità dei dispositivi medici rivolta al consumatore. Non occorre alcuna norma di raccordo: il meccanismo è quello dell’innesto diretto nella disciplina generale delle pratiche scorrette.
Le pratiche ingannevoli rilevanti per il settore dei dispositivi medici
Il dlgs 30/2026 recepisce la Direttiva (UE) 2024/825 modificando in modo organico il Codice del Consumo. Senza poter in questa sede analizzare nel dettaglio tutte le novità introdotte dal decreto, ci concentreremo invece sulle disposizioni che presentano il maggiore impatto per il settore dei dispositivi medici.
Più precisamente il nuovo decreto modifica alcune norme del codice del consumo i cui nuovi contenuti vanno tenuto in considerazione anche nel settore medical device.
Le nuove definizioni (articolo 18 Codice del Consumo)
L’articolo 18, comma 1, del Codice del Consumo viene arricchito con nuove definizioni. Le più rilevanti per il settore medtech sono le seguenti.
La lett. n-quater definisce l’“asserzione ambientale” come qualsiasi messaggio o rappresentazione, avente carattere non obbligatorio, che asserisce o implica che un dato prodotto, marca o operatore economico ha un impatto positivo o nullo sull’ambiente, oppure è meno dannoso rispetto ad altri, oppure ha migliorato il proprio impatto nel tempo. La definizione è amplissima: comprende testi, rappresentazioni figurative, grafiche, simboliche, marchi e nomi di prodotti. Per un fabbricante di dispositivi medici, questo significa che ogni comunicazione ambientale associata al prodotto – sul packaging, nel catalogo, sul sito web, nella brochure commerciale – è potenzialmente soggetta alla nuova disciplina.
La lett. n-quinquies introduce l’“asserzione ambientale generica”: un’asserzione la cui specificazione non è fornita in termini chiari ed evidenti tramite lo stesso mezzo di comunicazione. È il caso tipico del claim “ecologico” o “sostenibile” stampato sull’etichetta di un dispositivo senza alcun approfondimento verificabile.
Le lettere n-sexies e n-septies definiscono rispettivamente l’“etichetta di sostenibilità” e il “sistema di certificazione”, completando un vocabolario normativo che prima mancava del tutto.
Le pratiche in ogni caso ingannevoli: la nuova lista nera (articolo 23 Codice del Consumo)
La modifica dell’articolo 23 – le pratiche considerate in ogni caso ingannevoli, senza necessità di valutazione caso per caso – è forse quella che ha l’impatto pratico più immediato per il settore medtech. Il decreto aggiunge sei nuove lettere al comma 1.
La lett. b-bis vieta l’esibizione di un’etichetta di sostenibilità non basata su un sistema di certificazione o non stabilita da autorità pubbliche. Niente più auto-certificazioni ambientali: ogni etichetta che un’impresa appone su un dispositivo medico deve essere supportata da una verifica indipendente.
La lett. d-bis colpisce le asserzioni ambientali generiche per le quali il professionista non sia in grado di dimostrare l’eccellenza riconosciuta delle prestazioni ambientali. Chi scrive “eco-friendly” sull’etichetta di una siringa o di un kit diagnostico deve poter dimostrare la conformità al Regolamento Ecolabel UE (Regolamento (CE) numero 66/2010) o a un sistema equivalente conforme alla norma EN ISO 14024.
La lett. d-ter vieta di formulare un’asserzione ambientale concernente il prodotto nel suo complesso, o l’attività del professionista nel suo complesso, quando in realtà riguarda soltanto un determinato aspetto.
È il caso, assai frequente nel mondo dei dispositivi medici, del fabbricante che comunica la “sostenibilità” dell’intero dispositivo quando in realtà il miglioramento riguarda solo il packaging.
La lett. d-quater – e questa è, a parere di chi scrive, una delle norme più incisive – vieta di asserire che un prodotto abbia un impatto neutro, ridotto o positivo sull’ambiente in termini di emissioni di gas a effetto serra quando tale asserzione è basata esclusivamente sulla compensazione (offset) delle emissioni. I claim del tipo “impatto zero” o “climate neutral” fondati solo su crediti di carbonio sono ora espressamente vietati.
La lett. l-bis vieta, infine, di presentare requisiti imposti per legge come se fossero un tratto distintivo dell’offerta. La pratica – purtroppo diffusa anche nel settore dei dispositivi medici – di vantare come “esclusivi” obblighi che in realtà gravano su tutti gli operatori è ora espressamente sanzionata.
Le azioni ingannevoli rafforzate (articolo 21 Codice del Consumo)
L’articolo 21 del Codice del Consumo viene a sua volta modificato. Sul piano delle azioni ingannevoli, le “caratteristiche ambientali o sociali” e gli “aspetti relativi alla circolarità, quali la durabilità, la riparabilità o la riciclabilità” vengono inseriti tra le caratteristiche principali del prodotto suscettibili di ingannare il consumatore.
Soprattutto, la nuova lett. b-ter del comma 2 colpisce le asserzioni ambientali relative a prestazioni future prive di un piano di attuazione dettagliato, realistico, con obiettivi misurabili e scadenze precise, verificato periodicamente da un terzo indipendente. In sostanza: chi promette “carbon neutral entro il 2030” deve avere un piano credibile, con numeri, date e risorse, e deve farselo certificare.
Esempi concreti: i rischi di greenwashing
Per rendere operativo il quadro fin qui delineato, abbiamo provato ad immaginare alcuni scenari tipici di rischio. Gli esempi che seguono sono di natura meramente illustrativa e frutto di elaborazione, ma riflettono dinamiche reali e ricorrenti nella comunicazione commerciale del settore medtech.
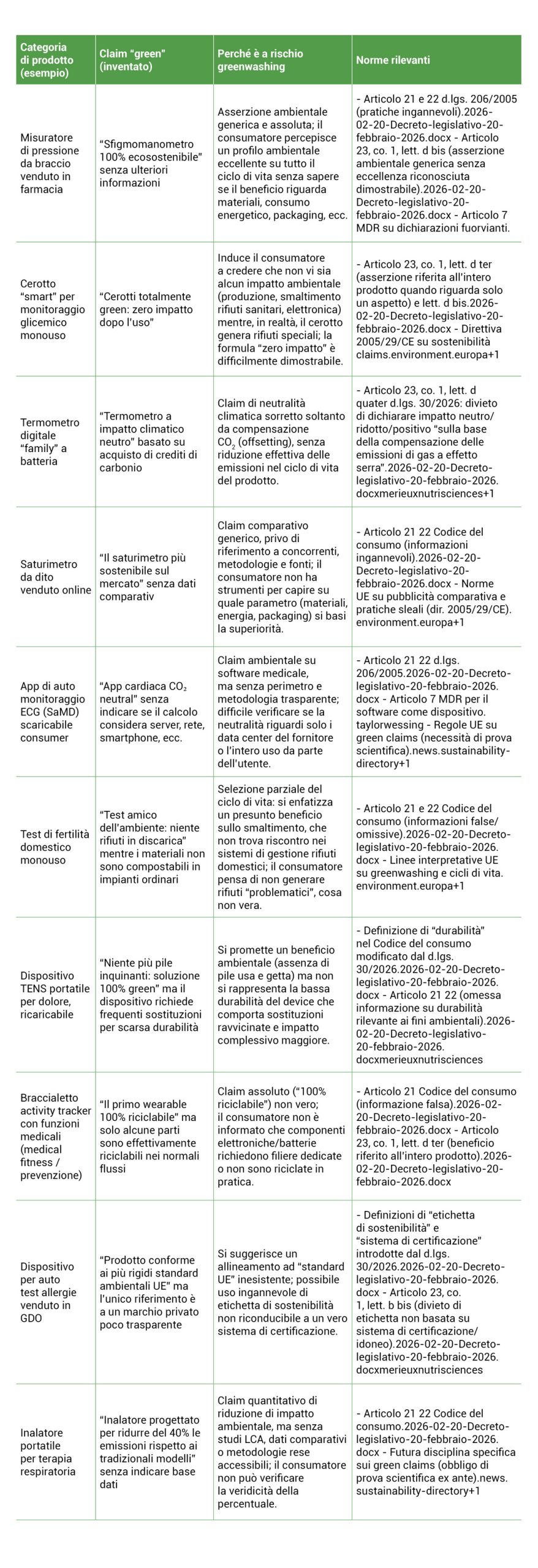
*Un elemento trasversale merita di essere evidenziato. In ciascuno degli esempi riportati, il rischio di greenwashing si intreccia con il rischio di violazione dell’articolo 7 MDR. Quando un claim ambientale incide sulla percezione delle prestazioni, della sicurezza o della destinazione d’uso di un dispositivo medico, il fabbricante si espone a una doppia contestazione: quella dell’AGCM ai sensi del Codice del Consumo (o del D.Lgs. 145/2007 per la comunicazione B2B) e quella del Ministero della Salute ai sensi della normativa settoriale.
Conclusioni: cosa devono fare ora le imprese del settore medtech
Il dlgs 30/2026 diventa pienamente efficace il 27 settembre 2026. Non è una norma pensata specificamente per il settore dei dispositivi medici, ma le sue conseguenze per questo settore possono essere tutt’altro che marginali. I fabbricanti, i distributori e gli importatori di dispositivi medici che comunicano caratteristiche ambientali dei propri prodotti – e sono sempre più numerosi – devono oggi confrontarsi con un quadro sanzionatorio significativamente più stringente. Vediamo allora cosa fare:
- Audit dei green claim esistenti. Ogni asserzione ambientale presente su prodotti, packaging, siti web, cataloghi, materiali di marketing e contratti deve essere mappata e verificata. Per ciascun claim occorre accertare che sia specifico (non generico), verificabile con dati oggettivi, riferito all’effettivo ambito di applicazione (prodotto, linea o intera azienda) e non basato esclusivamente su meccanismi di compensazione.
- Verifica delle certificazioni. Le etichette di sostenibilità devono essere basate su sistemi di certificazione sottoposti a controlli da parte di terzi indipendenti (articolo 23, comma 1, lett. b-bis, Codice del Consumo come modificato). Le auto-certificazioni prive di verifica esterna sono vietate.
- Estensione della revisione alla comunicazione B2B. Alla luce dell’applicazione per analogia dei nuovi criteri anche nell’ambito del D.Lgs. 145/2007, la revisione dei green claim non può limitarsi alla comunicazione al consumatore: deve riguardare anche i cataloghi, le presentazioni agli operatori sanitari, i materiali per gare d’appalto e ogni altra forma di comunicazione commerciale.
- Coordinamento con l’articolo 7 MDR. I green claim che incidano sulla percezione delle prestazioni, della sicurezza o della destinazione d’uso del dispositivo devono essere valutati anche alla luce dell’articolo 7 MDR. Il coordinamento tra normativa ambientale e normativa di settore è un aspetto che il decreto non affronta esplicitamente, ma che le imprese non possono permettersi di ignorare.
- Le funzioni aziendali coinvolte – marketing, comunicazione, regulatory affairs, legal – devono essere formate sulle nuove regole entro il 27 settembre 2026, data a partire dalla quale le nuove previsioni saranno applicabili.
Il rischio di violazione della norma non è solo sanzionatorio. Un’impresa del settore medtech che subisca un provvedimento AGCM per greenwashing affronta conseguenze reputazionali gravi e prolungate, in un settore nel quale la credibilità scientifica è un asset fondamentale.
ARTICOLO PUBBLICATO SU ABOUTPHARMA
Rubrica "I DISPOSITIVI MEDICI TRA NORMATIVA E REGOLATORIO"
Leggi gli altri articoli presenti nella nostra rubrica in collaborazione con AboutPharma
